The ETE toolkit
A Python Environment for Tree Exploration
A Python Environment for Tree Exploration
ETE-NPR is a portable application providing a complete environment for the design and execution of phylogenomic workflows, including super-matrix and gene-tree reconstruction approaches. ETE-NPR is designed to cover all necessary steps from alignment reconstruction to the generation of trees and alignment images.
ETE-NPR aims at providing a complete phylogenetic reconstruction environment both for novel and expert users.
The program is built on top of the ETE toolkit and a bunch of specialized software, providing predefined workflows based on up-to-date phylogenetic procedures.
In addition, ETE-NPR implements a Nested Phylogenetic Reconstruction (NPR) methodology that allows to address the inference of large phylogenies as a recursive process in which the resolution of internal nodes is gradually refined from root to leaves in an automatic way.
Note that NPR capabilities are also available for the supermatrix approach, which allows to refine the concatenated alignment of each internal node and automatically increase the number of marker genes used to infer the topology of clades including closer species. Related: https://peerj.com/preprints/223/
Finally, ETE-NPR allows the use of meta-workflows, which consist of multiple workflows executed at once on the same input data using a single command line. A common meta-workflow consist of several aligners and tree inference programs tested on the same to data to evaluate differences among results.
update_npr command to check for recent upgrades.sh ete-start in the command line to get a shell within the ETE-NPR environment (first execution will download and setup the dependencies automatally).Execute the update_npr command to check for recent upgrades.
If you already have a running version of the software, execute the update_npr command to upgrade to its latest version.
The main executable program is npr, which allows to run gene-tree and super-matrix workflows with or without recursion.
A list of available workflows can be obtained by typing npr wl, and further help using npr -h
Because of its modular design, ETE-NPR is able to stop and resume the execution of workflows. This has several consequences:
If you want to restart an analysis from scratch, you will need
to use a different output directory or use the --clearall option to delete or previous data.
When the execution of ETE-NPR is abruptly terminated (i.e. with Control-C), running processes are killed, but result from finished jobs will still be present in the output directory. Running the same command again will reuse finished data and restart missing jobs.
Shared tasks among workflows will be reused if the same output directory is used to run a different workflow on the same input data (even after finished executions) .
Multithreading options can be activated at any momment using the --cpu option.
The generation of tree and alignment images require a X11 environment in your system. To disable this option use the --noimg flag.
For the next examples, you will need to download the following data files:
Execute a fast analysis:
$ npr -a p53.fasta -o p53_results1 -w standard_fasttree
After a short time, your tree should be found in p53_results/, including (if graphics was available in your system) an image of your tree and alignment (Figure 2).
Workflows are specified using the -w
parameter, and several strategies can be applied at once, thus recycling common tasks.
$ npr -a p53.fasta -o p53_results -w standard_fasttree standard_raxml
Note that recursive phylogenies are only recommended for very large trees or very patchy alignments. Next examples are kept small to avoid large computation times.
Any workflow can be turned into recursive by ussing the --recursive option:
$ npr -a p53.fasta -o p53_results3 -w standard_fasttree --recursive
This command will execute the standard RAxML workflow for all the nodes in the resulting tree, meaning that alignment and model-selection steps will be optimized at each iteration.
As this can be very intensive computationally, you may want to specify different workflows for different tree levels.
$ npr -a p53.fasta -o p53_results4 -w standard_fasttree \
--recursive "size-range:0-19,standard_raxml" "size-range:20-500,standard_fasttree"
This command will execute the standard FastTree workflow for the first iteration, and for partitions of at least 20 sequences. Smaller partitions will be analyzed using RAxML.
Recursive phylogenies allow to combine the use of nucleotide and amino-acid sequnece in the same tree. You just need to specify a nucleotide fasta file and your recursive strategy
$ npr -a NUP62.aa.fa -n NUP62.nt.fa -o NUP62_results1 -w standard_fasttree \
--recursive seq-sim-range:0.95-1,standard_fasttree --nt_switch_thr 0.95
This command will execute the standard fasttree workflow using amino-acid sequences in the first iteration. Recursive refinement is done using the same workflow but will only occur in nodes whose sequence similarity (aa based) is higher than 0.95, which is also the threshold for switching to nucleotide alignments. Result is shown in Figure 3.

Fig 1. Standard output of a typical ETE-NPR
analysis. Different tasks are monitored during execution. The verbosity
of log messages can be controlled using the-v option.
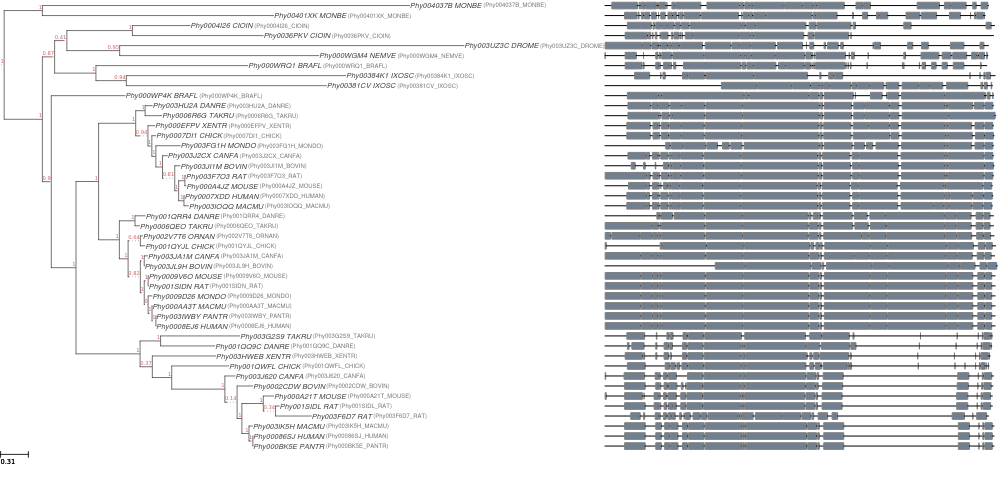
Fig 2. An image of the final tree generated by ETE-NPR, including the schematic representation of the multiple sequence alignment.
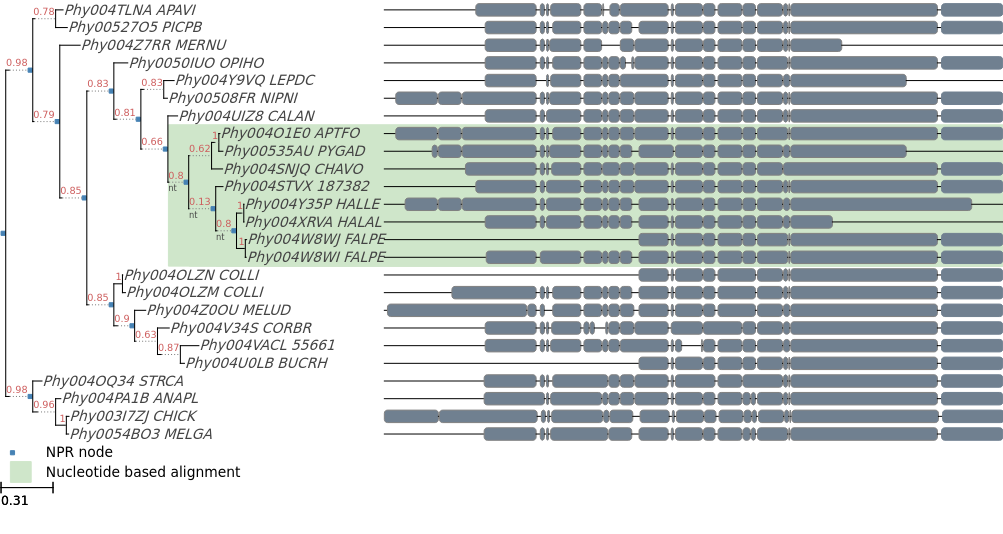
Fig 3. Blue nodes represent nodes whose internal topology has been optimized with NPR. Green background show the parts of the phylogeny that were optimized using a codon based alignment.
For the next examples, you will need to download the following data files:
super-matrix based strategies require:
The cogs file is text file containing a group of orthologous sequences per line, each sparated by TABs. In example:
sp1_seqA sp2_seqA sp3_seqA
sp1_seqB sp2_seqB sp3_seqB
sp1_seqC sp3_seqC sp4_seqC sp5_seqC
...
$ npr -a proteome_seqs.fa --cogs cogs.txt -o sptree1_results/ -m sptree_dummy -w linsi_fasttree
Check the (expanding) ETE-NPR HowTo
ETE is developed as an academic free software tool. If you find ETE useful for your work, please cite:
Jaime Huerta-Cepas, Joaquín Dopazo and Toni Gabaldón. ETE: a python Environment for Tree Exploration. BMC Bioinformatics 2010, 11:24. doi:10.1186/1471-2105-11-24
The ETE Toolkit was originally
developed at the bioinformatics
department of CIPF and greatly improved
at the comparative genomics unit
of CRG. At present, ETE is maintained by Jaime
Huerta-Cepas at the Structural and
Computational Biology unit of EMBL (Heidelberg,
Germany).![]()